Minimum Alignment Score Normalized to Read Length Galaxy
In this department nosotros will await at applied aspects of manipulation of side by side-generation sequencing information. Nosotros volition showtime with Fastq format produced by most sequencing machines and will finish with SAM/BAM format representing mapped reads.
Set your Galaxy to begin
- If y'all are new Galaxy → commencement with the Milky way 101 tutorial
- Create a new Milky way history at https://usegalaxy.org (don't forget to log in).
- Import the following iv datasets by cutting and pasting these URLs into Milky way's upload tool (for help see URL upload option in upload tutorial):
https://zenodo.org/record/583613/files/sample1-f.fq.gz https://zenodo.org/record/583613/files/sample1-r.fq.gz https://zenodo.org/tape/583613/files/sample2-f.fq.gz https://zenodo.org/record/583613/files/sample2-r.fq.gz when uploading these dataset set datatype to fastqsanger.gz. The animated prototype beneath shows the details of the entire upload process:
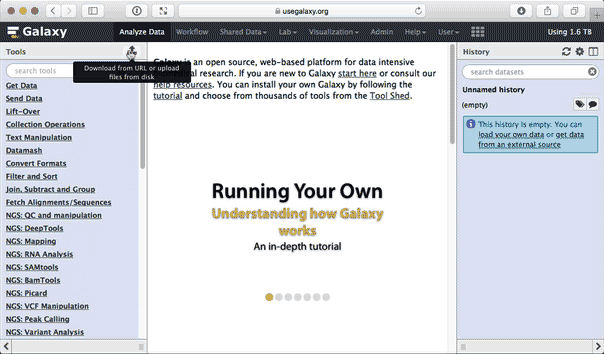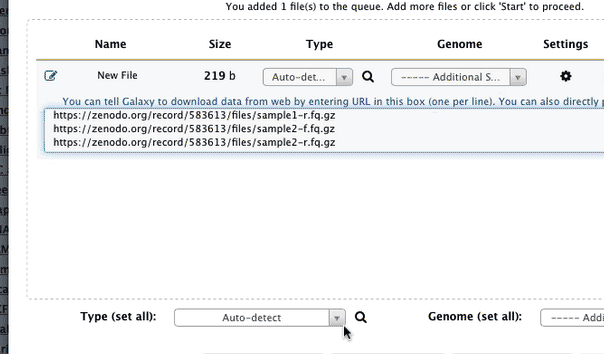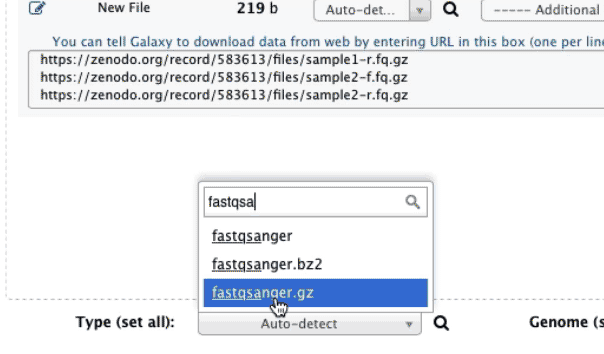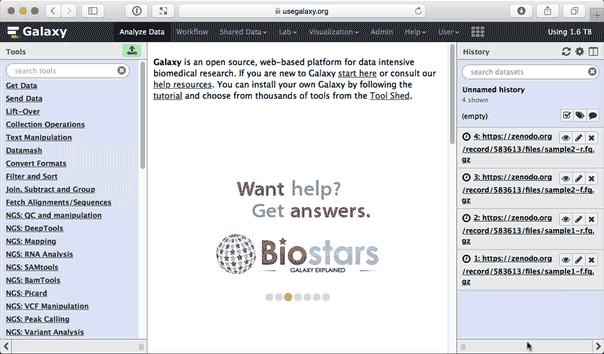 |
Figure 1. Uploading data from URL and setting datatype to fastqsanger.gz (this is a loop, so if you missed something information technology will repeat itself soon). |
These are paired stop data (datasets with -f is their filename are forward reads and datasets with -r are reverse) representing two contained sampled produced by an Illumina auto.
Upload the datasets. If you've done everything correctly, y'all will see Galaxy interface looking similar this:
 |
| Figure 2. Data are now uploaded. |
Fastq manipulation and quality command
What is Fastq?
FastQ is not a very well divers format. In the beginning various manufacturers of sequencing instruments were free to interpret fastq as they saw fit, resulting in a multitude of fastq flavors. This variation stemmed primarily from different means of encoding quality values as described here (below you volition find explanation of quality scores and their pregnant). Today, fastq Sanger version of the format is considered to be the standard form of fastq. Galaxy is using fastq sanger as the but legitimate input for downstream processing tools and provides a number of utilities for converting fastq files into this form (see NGS: QC and manipulation section of Galaxy tools).
Fastq format looks similar this:
@M02286:19:000000000-AA549:ane:1101:12677:1273 1:North:0:23 CCTACGGGTGGCAGCAGTGAGGAATATTGGTCAATGGACGGAAGTCTGAACCAGCCAAGTAGCGTGCAG + ABC8C,:@F:CE8,B-,C,-vi-9-C,CE9-CC--C-<-C++,,+;CE<,,CD,CEFC,@E9<FCFCF?9 @M02286:19:000000000-AA549:1:1101:15048:1299 1:N:0:23 CCTACGGGTGGCTGCAGTGAGGAATATTGGACAATGGTCGGAAGACTGATCCAGCCATGCCGCGTGCAG + ABC@CC77CFCEG;F9<F89<ix--C,CE,--C-6C-,CE:++7:,CF<,CEF,CFGGD8FFCFCFEGCF @M02286:xix:000000000-AA549:ane:1101:11116:1322 i:N:0:23 CCTACGGGAGGCAGCAGTAGGGAATCTTCGGCAATGGACGGAAGTCTGACCGAGCAACGCCGCGTGAGT + AAC<CCF+@@>CC,C9,F9C9@nine-CFFFE@vii@:+CC8-C@:7,@EFE,6CF:+8F7EFEEF@EGGGEEE Each sequencing read is represented past four lines:
-
@followed by read ID and optional information about sequencing run - sequenced bases
-
+(optionally followed by the read ID and some additional info) - quality scores for each base of the sequence encoded every bit ASCII symbols
Paired end data
Information technology is mutual to prepare pair-end and mate-pair sequencing libraries. This is highly beneficial for a number of applications discussed in subsequent topics. This is considering in addition to sequence data we know that forrad and contrary reads are physically linked inside the sequenced molecule. For now let'south just briefly hash out what these are and how they manifest themselves in fastq form.
 |
| Figure 3. Paired-end and mate-pair reads. In paired stop sequencing (left) the actual ends of rather brusque Deoxyribonucleic acid molecules (less than 1kb) are determined, while for mate pair sequencing (right) the ends of long molecules are joined and prepared in special sequencing libraries. In these mate pair protocols, the ends of long, size-selected molecules are connected with an internal adapter sequence (i.eastward. linker, yellow) in a circularization reaction. The round molecule is then processed using brake enzymes or fragmentation. Fragments are enriched for the linker and outer library adapters are added around the ii combined molecule ends. The internal adapter can then be used as a second priming site for an boosted sequencing reaction in the same orientation or sequencing tin exist performed from the second adapter, from the reverse strand. (From Ph.D. dissertation by Martin Kircher) |
Thus in both cases (paired-end and mate-pair) a unmarried physical piece of Dna (or RNA in the case of RNA-seq) is sequenced from ii ends and so generates two reads. These can be represented every bit divide files (two fastq files with first and second reads) or a single file were reads for each finish are interleaved. Here are examples:
Two unmarried files
File ane
@M02286:nineteen:000000000-AA549:1:1101:12677:1273 1:N:0:23 CCTACGGGTGGCAGCAGTGAGGAATATTGGTCAATGGACGGAAGTCT + ABC8C,:@F:CE8,B-,C,-6-ix-C,CE9-CC--C-<-C++,,+;CE @M02286:19:000000000-AA549:one:1101:15048:1299 ane:N:0:23 CCTACGGGTGGCTGCAGTGAGGAATATTGGACAATGGTCGGAAGACT + ABC@CC77CFCEG;F9<F89<nine--C,CE,--C-6C-,CE:++vii:,CF File 2
@M02286:19:000000000-AA549:1:1101:12677:1273 2:North:0:23 CACTACCCGTGTATCTAATCCTGTTTGATACCCGCACCTTCGAGCTTA + --8A,CCE+,,;,<CC,,<CE@,CFD,,C,CFF+@+@CCEF,,,B+C, @M02286:19:000000000-AA549:1:1101:15048:1299 2:N:0:23 CACTACCGGGGTATCTAATCCTGTTCGCTCCCCACGCTTTCGTCCATC + -6AC,EE@::CF7CFF<<FFGGDFFF,@FGGGG?F7FEGGGDEFF>FF Interleaved file
@1/1 AGGGATGTGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTA + EGGEGGGDFGEEEAEECGDEGGFEEGEFGBEEDDECFEFDD@CDD<ED @one/two CCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAAC + GHHHDFDFGFGEGFBGEGGEGEGGGHGFGHFHFHHHHHHHEF?EFEFF @2/ane AGGGATGTGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTA + HHHHHHEGFHEEFEEHEEHHGGEGGGGEFGFGGGGHHHHFBEEEEEFG @2/ii CCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAAC + HHHHHHHHHHHHHGHHHHHHGHHHHHHHHHHHFHHHFHHHHHHHHHHH Here the start and the 2d reads are identified with /1 and /ii tags (simply even without these tags we know that odd reads are in forwards orientation and even are in reverse).
What are base of operations qualities?
Equally we've seen above, fastq datasets comprise two types of information:
- sequence of the read
- base qualities for each nucleotide in the read.
The base qualities permit u.s.a. to approximate how trustworthy each base in a sequencing read is. The following excerpt from an fantabulous tutorial by Friederike Dündar, Luce Skrabanek, Paul Zumbo explains what base qualities are:
Illumina sequencing is based on identifying the individual nucleotides by the fluorescence signal emitted upon their incorporation into the growing sequencing read. In one case the fluorescence intensities are extracted and translated into the iv letter code. The deduction of nucleotide sequences from the images acquired during sequencing is unremarkably referred to equally base calling. Due to the imperfect nature of the sequencing process and limitations of the optical instruments, base calling volition always have inherent uncertainty. This is the reason why FASTQ files store the DNA sequence of each read together with a position-specific quality score that represents the error probability, i.e., how likely information technology is that an individual base phone call may be incorrect. The score is called Phred score, Q, which is proportional to the probability p that a base phone call is incorrect, where  . For instance, a Phred score of ten corresponds to one error in every ten base calls (
. For instance, a Phred score of ten corresponds to one error in every ten base calls ( ), or 90% accurateness; a Phred score of 20 corresponds to one error in every 100 base of operations calls, or 99% accuracy. A higher Phred score thus reflects higher conviction in the reported base. To assign each base a unique score identifier (instead of numbers of varying grapheme length), Phred scores are typically represented equally ASCII characters. At https://ascii-code.com/ you tin can run into which characters are assigned to what number. For raw reads, the range of scores will depend on the sequencing technology and the base caller used (Illumina, for example, used a tool called Bustard, or, more than recently, RTA). Unfortunately, Illumina has been anything only consequent in how they calculated and ASCII-encoded the Phred score (come across beneath)! In addition, Illumina now allows Phred scores for base calls with as loftier as 45, while 41 used to be the maximum score until the HiSeq Ten. This may cause issues with downstream applications that expect an upper limit of 41.
), or 90% accurateness; a Phred score of 20 corresponds to one error in every 100 base of operations calls, or 99% accuracy. A higher Phred score thus reflects higher conviction in the reported base. To assign each base a unique score identifier (instead of numbers of varying grapheme length), Phred scores are typically represented equally ASCII characters. At https://ascii-code.com/ you tin can run into which characters are assigned to what number. For raw reads, the range of scores will depend on the sequencing technology and the base caller used (Illumina, for example, used a tool called Bustard, or, more than recently, RTA). Unfortunately, Illumina has been anything only consequent in how they calculated and ASCII-encoded the Phred score (come across beneath)! In addition, Illumina now allows Phred scores for base calls with as loftier as 45, while 41 used to be the maximum score until the HiSeq Ten. This may cause issues with downstream applications that expect an upper limit of 41.

Base call qualities are represented using the Phred score scale. Dissimilar Illumina (formerly Solexa) versions used different scores and ASCII offsets. Starting with Illumina format i.8, the score at present represents the standard Sanger/Phred format that is also used by other sequencing platforms and the sequencing archives.
 |
| Figure 4. The ASCII interpretation and ranges of the dissimilar Phred score notations used past Illumina and the original Sanger estimation. Although the Sanger format allows a theoretical score of 93, raw sequencing reads typically exercise not exceed a Phred score of 60. In fact, near Illumina-based sequencing will upshot in maximum scores of 41 to 45 (image from Wikipedia). |
Assessing information quality
One of the get-go steps in the analysis of NGS data is seeing how good the information actually is. FastQC is a fantastic tool allowing y'all to evaluate the quality of fastq datasets (and deciding whether to arraign or not to arraign whoever has washed sequencing for you).
 |  |
| Figure 5. Left: Splendid quality. | Right: Hmmmm....Ok. |
Hither you tin come across FastQC base quality reports (the tools gives you many other types of data) for two datasets: A and B. The A dataset has long reads (250 bp) and very expert quality profile with no qualities dropping below phred score of thirty. The B dataset is significantly worse with ends of the reads dipping below phred score of 20. The B reads may need to exist trimmed for farther processing.
It may be challenging to use fastQC when you take a lot of datasets. For instance, in our case at that place are four datasets. FastQC needs to be run on each dataset individually and so one needs to await at each fastQC report individually. This may not exist a big trouble for four datasets, but information technology will go an issue if y'all take 100s or 1,000s of datasets. Phil Ewels has adult a tool called MultiQC that allows to summarize multiple QC reports at in one case. To run MultiQC yous need to run fastQC on private datasets and and so feed fastQC outputs to MultiQC (note that MultiQC is not express to processing FastQC reports but accepts outputs of many other tools). Galaxy makes this easy as shown in the following video:
In this video we run FastQC on the four datasets and so summarized these data with MultiQC. The post-obit figure shows one of the graphs produced past MultiQC:
 |
Figure vi. A MultiQC report showing quality score distribution for the iv sequences using in this tutorial. Here sample1-f has highest quality: its quality scores never dip below phred score of 25. The other datasets are slightly worse, only all are by and large acceptable. |
Trimming reads
One of the conclusions from our QC analyses (Fig. half-dozen) is that the quality is acceptable only drops towards the end of the reads (this is typical for Illumina which uses contrary terminators bases with cleave-able colour labels. Because the procedure of cleaving terminators and color labels is not 100% efficient racket accumulates as run progresses and and so bases at the ends of reads tend to have lower quality). There is a number of steps nosotros can take to mitigate the effect of low quality bases. Ane is dynamically trim the reads:
- slide a window across reads
- at every step of the process calculate boilerplate quality of bases within the given window
- if quality drops beneath certain set threshold → terminate and trim the read of the read from this betoken until the end
- output the beginning of the read
One of the tools that performs this procedure is Trimmomatic developed by the Usadel lab. Allow's utilise NGS: QC and manipulation → Trimmomatic to trim out four datasets:
 |
| Figure seven. Trimming our datasets with Trimmomatic. Hither reads will be trimmed if the base quality averaged beyond 4 bases drops beneath 25. |
To meet the issue of trimming on the reads let'south take Trimmomatic output, run it through FastQC (NGS: QC and manipulation → FastQCand summarize with MultiQC (NGS: QC and manipulation → multiQC). Below is the quality score distribution graph (the same graph shown in Fig. 6):
 |
| Effigy viii. Quality score distribution for trimmed datasets. Compare this image with Fig. half dozen. You tin see that sequences are shorted simply quality is significantly higher. |
We will now utilize trimmed reads every bit the input to downstream analyses.
Try it yourself
QC, trim, and QC again datasets you take uploaded earlier to produce a concluding set of sequences we volition be using downstream.
Mapping your information
Mapping of NGS reads against reference sequences is ane of the fundamental steps of the analysis. At present it is time to see how this is done in practise. Beneath is a list of fundamental publications highlighting mainstream mapping tools:
- 2009 Bowtie i - Langmead et al.
- 2012 Bowtie ii - Langmead and Salzberg
- 2009 BWA - Li and Durbin
- 2010 BWA - Li and Durbin
- 2013 BWA-MEM - Li
Mapping against a pre-computed genome alphabetize
Mappers unremarkably compare reads confronting a reference sequence that has been transformed into a highly accessible data construction called genome alphabetize. Such indexes should be generated before mapping begins. Galaxy instances typically store indexes for a number of publicly bachelor genome builds.
 |
| Figure ix. Mapping against a pre-computed alphabetize in Milky way. |
For example, the image above shows indexes for hg38 version of the man genome. You can encounter that there are actually three choices: (one) hg38, (2) hg38 canonical and (iii) hg38 canonical female. The hg38 contains all chromosomes also as all unplaced contigs. The hg38 approved does not contain unplaced sequences and only consists of chromosomes 1 through 22, 10, Y, and mitochondria. The hg38 canonical female contains everything from the canonical set with the exception of chromosome Y.
The post-obit video show mapping using BWA-MEM:
Try it yourself
Map datasets uploaded before using BWA against hg38 version of the human genome.
What if pre-computed index does not be?
If Galaxy does not accept a genome you lot need to map against, you can upload your genome sequence equally a FASTA file and use it in the mapper directly as shown below (Load reference genome is prepare to History).
 |
| Effigy x. Mapping confronting a custom index in Galaxy |
In this case Milky way will kickoff create an index from this dataset and and so run mapping analysis confronting it. The following video shows how this works in practice:
SAM/BAM datasets
The SAM/BAM format is an accepted standard for storing aligned reads (it tin also shop unaligned reads and some mappers such equally BWA are accepting unaligned BAM equally input). The binary form of the format (BAM) is compact and can be chop-chop searched (if indexed). In Galaxy BAM datasets are ever indexed (accompanies by a .bai file) and sorted in coordinate order. In the post-obit discussion I in one case again rely on tutorial by Friederike Dündar, Luce Skrabanek, and Paul Zumbo.
The Sequence Alignment/Map (SAM) format is a generic nucleotide alignment format that describes the alignment of sequencing reads (or query sequences) to a reference. The human readable, TAB-delimited SAM files can exist compressed into the Binary Alignment/Map format. These BAM files are bigger than just gzipped SAM files, because they accept been optimized for fast random admission rather than size reduction. Position-sorted BAM files tin can be indexed so that all reads aligning to a locus can be efficiently retrieved without loading the entire file into retentiveness.
As shown below, SAM files typically contain a header section and an alignment department where each row represents a single read alignment. The following sections volition explicate the SAM format in a bit more than detail. For the virtually comprehensive and updated data go to https://github.com/samtools/hts-specs.
 |
| Figure 11. Schematic representation of a SAM file. Each line of the optional header section starts with "@", followed by the appropriate abbreviation (due east.thousand., SQ for sequence lexicon which lists all chromosomes names (SN) and their lengths (LN)). The vast majority of lines within a SAM file typically represent to read alignments where each read is described by the 11 mandatory entries (black font) and a variable number of optional fields (grey font; from tutorial past Friederike Dündar, Luce Skrabanek, and Paul Zumbo). |
The header section includes data virtually how the alignment was generated and stored. All lines in the header section are tab-delimited and begin with the "@" character, followed by tag:value pairs, where tag is a two-letter of the alphabet string that defines the content and the format of value. For case, the "@SQ" line in the header section contains the data well-nigh the names and lengths of the *reference sequences to which the reads were aligned. For a hypothetical organism with 3 chromosomes of length one,000 bp, the SAM header should comprise the following 3 lines:
@SQ SN:chr1 LN:1000 @SQ SN:chr2 LN:yard @SQ SN:chr3 LN:thousand SAM alignment section
The optional header section is followed by the alignment section where each line corresponds to one sequenced read. For each read, there are xi mandatory fields that e'er appear in the aforementioned social club:
<QNAME> <FLAG> <RNAME> <POS> <MAPQ> <CIGAR> <MRNM> <MPOS> <ISIZE> <SEQ> <QUAL> If the corresponding information is unavailable or irrelevant, field values can be '0' or '*' (depending on the field, run across below), only they cannot be missing! Later on the 11 mandatory fields, a variable number of optional fields can be nowadays. Here's an example of one single line of a existent-life SAM file (you may need to scroll sideways):
ERR458493 .552967 xvi chrI 140 255 12 M61232N37M2S * 0 0 CCACTCGTTCACCAGGGCCGGCGGGCTGATCACTTTATCGTGCATCTTGGC BB?HHJJIGHHJIGIIJJIJGIJIJJIIIGHBJJJJJJHHHHFFDDDA1+B NH:i:i Hi:i:1 Equally:i:41 nM:i:2 The following table explains the format and content of each field. The FLAG, CIGAR, and the optional fields (marked in blue) are explained in more detail beneath. The number of optional fields can vary widely between different SAM files and even between reads within in the same file. The field types marked in blue are explained in more detail in the primary text below.

FLAG field
The FLAG field encodes various pieces of information about the individual read, which is especially important for PE reads. It contains an integer that is generated from a sequence of Boolean bits (0, 1). This style, answers to multiple binary (Aye/No) questions can be compactly stored as a serial of bits, where each of the unmarried bits can exist addressed and assigned separately.
The following tabular array gives an overview of the different properties that can be encoded in the FLAG field. The developers of the SAM format and samtools tend to apply the hexadecimal encoding equally a means to refer to the unlike bits in their documentation. The value of the FLAG field in a given SAM file, however, will always exist the decimal representation of the sum of the underlying binary values (as shown in Table below, row 2).
 |
Figure 12. The FLAG field of SAM files stores information near the respective read alignment in one single decimal number. The decimal number is the sum of all the answers to the Yes/No questions associated with each binary bit. The hexadecimal representation is used to refer to the individual bits (questions). A bit is fix if the respective state is true. For example, if a read is paired, 0x1 will be gear up, returning the decimal value of 1. Therefore, all FLAG values associated with paired reads must be uneven decimal numbers. Conversely, if the 0x1 bit is unset (= read is not paired), no assumptions can be made about 0x2, 0x8, 0x20, 0x40 and 0x80 because they refer to paired reads (from tutorial past Friederike Dündar, Luce Skrabanek, and Paul Zumbo). |
In a run with single reads, the flags you most commonly run across are:
- 0: This read has been mapped to the forrard strand. (None of the bit-wise flags accept been prepare.)
- 4: The read is unmapped (
0x4is ready). - 16: The read is mapped to the opposite strand (
0x10is set)
(0x100, 0x200 and 0x400 are non used by about aligners/mappers, simply could, in principle be set for single reads.) Some common FLAG values that you may come across in a PE experiment include:
| FLAG value | Meaning |
|---|---|
| 69 (= 1 + 4 + 64) | The read is paired, is the first read in the pair, and is unmapped. |
| 77 (= i + four + viii + 64) | The read is paired, is the get-go read in the pair, both are unmapped. |
| 83 (= 1 + 2 + 16 + 64) | The read is paired, mapped in a proper pair, is the commencement read in the pair, and it is mapped to the opposite strand. |
| 99 (= 1 + 2 + 32 + 64) | The read is paired, mapped in a proper pair, is the first read in the pair, and its mate is mapped to the reverse strand. |
| 133 (= one + four + 128) | The read is paired, is the second read in the pair, and it is unmapped. |
| 137 (= 1 + 8 + 128) | The read is paired, is the second read in the pair, and it is mapped while its mate is not. |
| 141 (= 1 + 4 + 8 + 128) | The read is paired, is the second read in the pair, simply both are unmapped. |
| 147 (= ane + 2 + 16 + 128) | The read is paired, mapped in a proper pair, is the second read in the pair, and mapped to the reverse strand. |
| 163 (= i + 2 + 32 + 128) | The read is paired, mapped in a proper pair, is the second read in the pair, and its mate is mapped to the reverse strand. |
A useful website for quickly translating the FLAG integers into plainly English explanations like the ones shown above is: https://broadinstitute.github.io/picard/explain-flags.html
CIGAR string
CIGAR stands for Curtailed Idiosyncratic Gapped Alignment Report. This sixth field of a SAM file contains a so-chosen CIGAR string indicating which operations were necessary to map the read to the reference sequence at that detail locus.
The post-obit operations are defined in CIGAR format (besides see figure below):
- M - Alignment (tin can be a sequence lucifer or mismatch!)
- I - Insertion in the read compared to the reference
- D - Deletion in the read compared to the reference
- Due north - Skipped region from the reference. For mRNA-to-genome alignments, an Due north functioning represents an intron. For other types of alignments, the interpretation of N is not defined.
- Southward - Soft clipping (clipped sequences are present in read); Southward may just accept H operations between them and the ends of the string
- H - Hard clipping (clipped sequences are NOT nowadays in the alignment record); can only be nowadays equally the commencement and/or concluding operation
- P - Padding (silent deletion from padded reference)
- = - Sequence match (not widely used)
- X - Sequence mismatch (not widely used)
The sum of lengths of the M, I, South, =, 10 operations must equal the length of the read. Hither are some examples:
 |
| Effigy 13. Examples of CIGAR strings (from tutorial by Friederike Dündar, Luce Skrabanek, and Paul Zumbo). |
Optional fields
Following the eleven mandatory SAM file fields, the optional fields are presented every bit key-value pairs in the format of <TAG>:<Blazon>:<VALUE>, where TYPE is one of:
-
A- Grapheme -
i- Integer -
f- Float number -
Z- String -
H- Hex string
The data stored in these optional fields volition vary widely depending on the mapper and new tags can exist added freely. In improver, reads within the aforementioned SAM file may have different numbers of optional fields, depending on the program that generated the SAM file. Commonly used optional tags include:
-
Every bit:i- Alignment score -
BC:Z- Barcode sequence -
Hello:i- Match is i-th hit to the read -
NH:i- Number of reported alignments for the query sequence -
NM:i- Edit distance of the query to the reference -
Doctor:Z- String that contains the exact positions of mismatches (should complement the CIGAR string) -
RG:Z- Read group (should lucifer the entry after ID if @RG is present in the header.
Thus, for example, we can use the NM:i:0 tag to select only those reads which map perfectly to the reference(i.e., have no mismatches). While the optional fields listed above are fairly standardized, tags that begin with X, Y, and Z are reserved for particularly free usage and will never be office of the official SAM file format specifications. XS, for case, is used by TopHat (an RNA-seq analysis tool we will discuss subsequently) to encode the strand information (e.yard., XS:A:+) while Bowtie2 and BWA use XS:i: for reads with multiple alignments to store the alignment score for the side by side-best-scoring alignment (e.g., XS:i:30).
Read Groups
Ane of the fundamental features of SAM/BAM format is the ability to label individual reads with readgroup tags. This allows pooling results of multiple experiments into a single BAM dataset. This significantly simplifies downstream logistics: instead of dealing with multiple datasets one can handle just one. Many downstream assay tools such as variant callers are designed to recognize readgroup data and output results on per-readgroup basis.
1 of the best descriptions of BAM readgroups is on GATK support site. We have gratefully stolen 2 tables describing the virtually important readgroup tags - ID, SM, LB, and PL - from GATK forum and provide them here:

GATK forum too provides the following case:

To see an example of read grouping manipulation in Galaxy come across the following video. In this video we use two BAM datasets as an example. We add readgroups and shown how information technology changes the underling BAM (SAM) information:
Effects of PCR duplicates
Preparation of sequencing libraries (at least at the fourth dimension of writing) for technologies such as Illumina involves PCR amplification. It is required to generate sufficient number of sequencing templates so that a reliable detection tin exist performed past base callers. Yet PCR has it'southward biases, which are peculiarly profound in cases of multitemplate PCR used for construction of sequencing libraries (Kanagawa et al. 2003).
 |
| Effigy xiv. Analyzing molecules aligning with the same outer coordinates, a mapping quality of at least 30 and a length of at least 30nt, resulted in an average coverage of 12.nine per PCR duplicate and an empirical coverage distribution similar to an exponential/power law distribution (left upper console). This indicates that many molecules are only observed for deeper sequencing while other molecules are available at higher frequencies. Analyzing length (left middle panel) and GC content (left lower console) patterns as well as the combination (right panel) shows higher PCR duplicate counts for a GC content between 30% to seventy% as well every bit for shorter molecules compared to longer molecules. This effect may exist due to an amplification bias from the polymerase or the cluster generation procedure necessary for Illumina sequencing. From Ph.D. dissertation of Martin Kircher). |
Duplicates can be identified based on their outer alignment coordinates or using sequence-based clustering. One of the mutual ways for identification of indistinguishable reads is the MarkDuplicates utility from Picard package. Information technology is designed to identify both PCR and optical duplicates:
Duplicates are identified as read pairs having identical 5' positions (coordinate and strand) for both reads in a mate pair (and optionally, matching unique molecular identifier reads; see BARCODE_TAG option). Optical, or more broadly Sequencing, duplicates are duplicates that appear amassed together spatially during sequencing and tin ascend from optical/imagine-processing artifacts or from bio-chemic processes during clonal amplification and sequencing; they are identified using the READ_NAME_REGEX and the OPTICAL_DUPLICATE_PIXEL_DISTANCE options. The tool's main output is a new SAM or BAM file in which duplicates take been identified in the SAM flags field, or optionally removed (see REMOVE_DUPLICATE and REMOVE_SEQUENCING_DUPLICATES), and optionally marked with a duplicate type in the 'DT' optional attribute. In improver, it also outputs a metrics file containing the numbers of READ_PAIRS_EXAMINED, UNMAPPED_READS, UNPAIRED_READS, UNPAIRED_READ DUPLICATES, READ_PAIR_DUPLICATES, and READ_PAIR_OPTICAL_DUPLICATES. Usage instance: coffee -jar picard.jar MarkDuplicates I=input.bam \ O=marked_duplicates.bam Thou=marked_dup_metrics.txt.
Sampling coincidence duplicates
Nevertheless, one has to be careful when removing duplicates in cases when the sequencing targets are small (e.g., sequencing of bacterial, viral, or organellar genomes besides equally amplicons). This is because when sequencing target is small reads volition have the same coordinates past adventure and non because of PCR distension issues. The figure below illustrates the fine balance between estimates allele frequency, coverage, and variation in insert size:
 |
| Figure 15. The Variant Allele Frequency (VAF) bias determined by coverage and insert size variance. Reads are paired-end and read length is 76. The insert size distribution is modeled equally a Gaussian distribution with mean at 200 and standard deviation shown on the ten-axis. The true VAF is 0.05. The darkness at each position indicates the magnitude of the bias in the VAF. (From Zhou et al. 2013). |
Putting it all together
In Galaxy we support four major toolsets for processing of SAM/BAM datasets:
- DeepTools - a suite of user-friendly tools for the visualization, quality control and normalization of data from deep-sequencing DNA sequencing experiments.
- SAMtools - diverse utilities for manipulating alignments in the SAM/BAM format, including sorting, merging, indexing and generating alignments in a per-position format.
- BAMtools - a toolkit for reading, writing, and manipulating BAM (genome alignment) files.
- Picard - a set of Java tools for manipulating high-throughput sequencing data (HTS) data and formats.
The following 2 videos highlight major steps of fastq-to-BAM analysis trajectory.
Organizing and QC'ing multiple datasets
In a typical analysis scenario a user usually processes multiple datasets. To brand the two following videos representative of existent-alive analyses we use a prepare of four samples, each consisting of ii forward and contrary sets of reads for a full of eight fastq datasets. The first video describes upload, QC, and preparation of these datasets for the subsequent analysis. The effigy below outlines steps highlighted in the video:
 |
Figure sixteen. The analysis shown in this figure and the following video begins with uploading of 8 datasets into history. These datasets are commencement combined into a flat drove - a unmarried entity containing eight fastq datasets. This drove is then analyzed with fastQC tool. This analyses produces another drove containing 8 fastQC outputs. Because it is inconvenient to look at private fastQC reports, we feed the entire collection to multiQC tool, which produces a single summary outputs accumulation data from 8 fastQC reports. At this point nosotros are happy with the quality of the data and prepare to motion on with the subsequent analysis. To do this we organize our datasets into a different type of collection - a paired collection. You can encounter that the paired collection is "deeper" that a flat drove: it contains samples and for each sample is lists respective sets of forward and contrary reads (shown and red and blue boxes with "F" and "R"). To learn more than nearly collections see this tutorial. |
After QC'ing nosotros motion on to map the reads, process the resulting BAM datasets, and visualize coverage in a genome browser. The post-obit figure and video detail these steps:
 |
Figure 17. In this assay (come across video below) we begin with a paired collection of fastq datasets. Mapping this collection to human genome with bwa mem produces a flat collection of BAM datasets. (EXTREMELY Of import: when mapping with bwa mem nosotros gear up readgroups (at time mark 00:40 in the video). This allows us to merge individual BAM datasets into one at the end of this assay.) Next using Picard'due south MarkDuplicates tool we process output of bwa mem. This pace produces two collections: (i) a collection of deduplicated BAMs and (two) a drove of indistinguishable metrics data produced by MarkDuplicates tool. We employ multiQC to visualize the duplicate metrics. Nosotros and so filter BAM drove produced by MarkDuplicates using Filter SAM or BAM tool to retain only properly mapped reads with mapping quality above 20 and mapping only to mitochondria (chrM). Finally output of the filtering step is merged with MergeSAM tool and displayed in the UCSC Genome Browser. Once more, merging is simply possible because we take set the readgroups during the mapping step. |
The merged BAM file can be using in a multifariousness of downstream analyses. In this case we simply used four samples represented by 8 paired fastq datasets. Drove, prominently featured in this tutorial, go far like shooting fish in a barrel to apply the same verbal analysis logic to 100s or ane,000s of datasets.
Effort it yourself
Perform a similar analyses with your own information.
If things don't work...
- ...create an issue by clicking "New issue" push button hither
- ...mutter. Employ Galaxy's support forum to do this.
Source: https://galaxyproject.org/tutorials/ngs/
0 Response to "Minimum Alignment Score Normalized to Read Length Galaxy"
Post a Comment